RNA Analysis
Structural RNAs:
A. Ribosomal RNA analysis
StructRNAfinder
StructRNAfinder
predicts and annotates RNA families in transcript or genome sequences.
This single tool not only displays the sequence/structural consensus
alignments for each RNA family, according to Rfam database but also
provides a taxonomic overview for each assigned functional RNA.
(Reference: Arias-Carrasco R et al. (2018) 19: 55).
Silva
Silva rRNA database project (Max Planck Institute for Marine Microbiology, Bremen, Germany ) - provides comprehensive, quality checked and regularly updated datasets of aligned small (16S/18S, SSU) and large subunit (23S/28S, LSU) ribosomal RNA (rRNA) sequences for all three domains of life (Bacteria, Archaea and Eukarya).
B. Transfer RNAs (tRNA)
For additional information see the Genomic tRNA Database GtRNAdb (Reference: Chan PP & Lowe TM (2016) Nucleic Acids Res; 44(D1): D184-189), or tRNADB-CE (tRNA Gene DataBase Curated by Experts) (Reference: Abe T et al. (2014) Front Genetics 5: 114).
tRNAscan-SE
tRNAs:
tRNAscan-SE
(Univerisity of California, Santa Cruz, U.S.A.) - is incredibly sensitive
& also provides secondary structure diagrams of the tRNA molecules
(Reference: Lowe, TM, & Eddy, SR. Nucleic Acids Res.
1997. 25: 955-964).
tRNAscan-SE 2.0 can also be accessed as part of the RNA Galaxy workbench 2.0
and stand alone here
ARAGORN
ARAGORN - employs heuristic algorithms to predict tRNA
secondary structure, based on homology with recognized tRNA consensus sequences and ability to form a base-paired
cloverleaf. tmRNA genes are identified using a modified version of the BRUCE program.
(Reference: Laslett, D. & Canback. 2004. Nucleic
Acids Research 32:11-16),
tRNAfinder
tRNAfinder - tRNA candidates are
detected based on tRNA cloverleaf secondary structure. Secondly, tRNAs are selected from the candidates based on the
consensus rules derived from real tRNAs.
(Reference: Kinouchi M & Kurokawa K (2006) J Computer Aided Chem 7: 116-126).
Rfam
Rfam - The Rfam database
is a collection of RNA families, each represented by multiple sequence
alignments, consensus secondary structures and covariance models
(Reference: Gardner, P.P. et al. 2008. Nucl. Acids
Res. 37, Database issue D136-D140)
C. Micro RNAs
(miRNAs) are small, non-coding RNA (~20-22 nucleotides) that negatively regulate gene expression at post-transcriptional level. You might want to start with miRGator3. (Reference: Cho S et al. (2013) Nucleic Acids Res 41(Database issue): D252-257).
mirTools 2.0
mirTools 2.0
-is an updated version of mirTools 1.0, which includes the following new
features. (1) From miRNA discovery in mirTools 1.0, mirTools 2.0 allows
users to detect and profile various types of ncRNAs, such as miRNA, tRNA,
snRNA, snoRNA, rRNA, and piRNA. (2) From miRNA profiling in mirTools 1.0,
mirTools 2.0 allows users to identify miRNA-targeted genes and performs
detailed functional annotation of miRNA targets, including Gene Ontology,
KEGG pathway and protein-protein interaction. (3) From comparison of two
samples for differentially expressed miRNAs in mirTools 1.0, mirTools 2.0
allows users to detect differentially expressed ncRNAs between two
experimental groups or among multiple samples. (4) Other significant
improvements include strategies used to detect novel miRNAs and piRNAs,
more taxonomy categories to discover more known miRNAs and a stand-alone
version of mirTools 2.0.
(Reference: Wu Jet al. (2013) RNA Biol; 10(7):
1087-1092).
miRDB
miRDB - employs an
improved algorithm for miRNA target prediction, we now present updated
transcriptome-wide target prediction data in miRDB, including 3.5 million
predicted targets regulated by 7000 miRNAs in five species. Further, we
have implemented the new prediction algorithm into a web server, allowing
custom target prediction with user-provided sequences. Another new
database feature is the prediction of cell-specific miRNA targets. miRDB
now hosts the expression profiles of over 1000 cell lines and presents
target prediction data that are tailored for specific cell models.
(Reference: Chen Y & Wang X (2020) Nucleic Acids Res
48(D1): D127-D131).
MirGeneDB 3.0
MirGeneDB 3.0 is a
database of manually curated microRNA genes that have been validated and
annotated. MirGeneDB 3.0 includes more than 21,000 microRNA gene entries
representing more than 1,700 microRNA families from 114 metazoan species.
All microRNAs can be browsed, searched and downloaded
(Reference: Clarke AW et al. (2025) Nucleic Acids
Res 53(D1): D116-D128).
miRNAFold
miRNAFold
- is a web server for fast miRNA precursor prediction in genomes that
allows predicting miRNA hairpin structures quickly with high sensitivity.
(Reference: Tav C et al (2016). Nucleic Acids
Research 44(W1): W181-W184)
miRViz
miRViz - is a
webserver application designed to visualize and interpret large miRNA
datasets, with no need for programming skills. MiRViz has two main goals:
(i) to help biologists to raise data-driven hypotheses and (ii) to share
miRNA datasets in a straightforward way through publishable quality data
representation, with emphasis on relevant groups of miRNAs. MiRViz can
currently handle datasets from 11 eukaryotic species
(Reference: Giroux C et al. (2020) Nucleic Acids Res
48(W1): W252-W261).
miR-BAG
miR-BAG predict miRNAs from the genomic sequences as well
as from Next Generation Sequencing data. It applies a bootstrap
aggregating approach to create an ensemble of three different approaches
(naïve Bayes, Best First Decision tree and SVM) to achieve a high
accuracy. At present miR-BAG includes 6 different species, 4 for animals
(Homo sapiens, Canis familiaris, Mus musculus, Rattus norvegicus)
alongwith one nematode (Caenorhabditis elegans) and one insect species
(Drosophila melanogaster). miR-BAG was found to perform consistently with
accuracy level higher than 90% for several species.
(Reference: Jha, A. et al. 2012. PLoS ONE 7(9):
e45782.)
Small nucleolar RNAs (snoRNAs)
Small nucleolar RNAs (snoRNAs) - can be detected with
Snoscan
for methylation-guide for snoRNAs and
snoGPS
for pseudouridylation-guide snoRNAs
(Reference: P. Schattner et al. 2005. Nucl. Acids
Res. 33: W686-W689).
Test sequences.
sRNAtoolbox
sRNAtoolbox
- is an integrated collection of small RNA research tools. Includes:
sRNAbench: Expression profiling of small RNAs and prediction of novel
microRNAs from deep sequencing data; sRNAde: Differential expression
analysis; sRNAblast: Blast analysis of deep sequencing reads against a
local nt/nr (NCBI link) database.
(Reference: A. Rueda et al. 2015. Nucl. Acids Res.
43 (W1): W467-W473).
D. Non-coding RNAs:
R2DT
R2DT
- Non-coding RNAs (ncRNA) are essential for all life, and their functions often depend on their secondary (2D) and tertiary structure. Despite the abundance of software for the visualisation of ncRNAs, few automatically generate consistent and recognisable 2D layouts, which makes it challenging for users to construct, compare and analyse structures. R2DT is a method for predicting and visualising a wide range of RNA structures in standardised layouts.
(Reference: Sweeney BA et al. 2021. Nature Communications 12: 3494).
E. Others - Meta site:
StructRNAfinder
StructRNAfinder -
is an automated tool for the identification, functional annotation and taxonomic assignation of RNA families through
secondary structure inference. These include: non-coding RNA genes (Gene) are composed by bona-fide RNAs with a recognised
function (e.g. CRISPR, miRNAs, ribozymes, rRNAs, snoRNAs); structured cis-regulatory elements (Cis-reg), are represented by
structural regulatory motifs available in RNA sequences (e.g. frameshift elements, riboswitches, thermoregulators); and Intron,
composed by self-splicing RNAs. It integrates third-part softwares to compare nucleotide sequences with sequence/structure
covariance models, generate secondary structures and functional annotations, in a webserver and stand-alone toolkit with
friendly reports and outputs useful for downstream analysis and data exploration.
(Reference: Arias-Carrasco R et al. 2018. BMC Bioinformatics, 19(1): 55).
RNA folding:
RNAstructure
RNAstructure
(webservers for RNA secondary structure prediction) is a software package
that includes structure prediction by free energy minimization, prediction
of base pairing probabilities, prediction of structures composed of highly
probably base pairs, and prediction of structures with pseudoknots.
(Reference: Xu ZZ & Mathews DH (2016) Methods Mol
Biol; 1490: 15-34).
RNAfold
RNAfold - this web server will predict secondary structures of single stranded RNA or DNA sequences. Current limits are 7,500 nt for partition function calculations and 10,000 nt for minimum free energy only predictions.
Vfold Pipeline
Vfold Pipeline
- offers a new user-friendly approach to the fully automated prediction of
RNA 3D structures with given sequences. It first predicts 2D structures
using the Vfold2D model and then predicts 3D structures based on the
predicted 2D structures using the Vfold3D and VfoldLA models.
(Reference: Xu, XJ., Chen, S.-J. (2016) Methods Mol
Biol. 1490: 63-72).
LocARNA
LocARNA
- Multiple Alignment of RNAs - is a tool for multiple alignment of RNA
molecules. LocARNA requires only RNA sequences as input and will
simultaneously fold and align the input sequences. LocARNA outputs a
multiple alignment together with a consensus structure. For the folding it
makes use of a very realistic energy model for RNAs as it is by RNAfold
of the Vienna RNA package (or Zuker's mfold). For the alignment it
features RIBOSUM-like similarity scoring and realistic gap cost.
(Reference: C. Smith et al. 2010. Nucl. Acids Res.
38: W373-377).
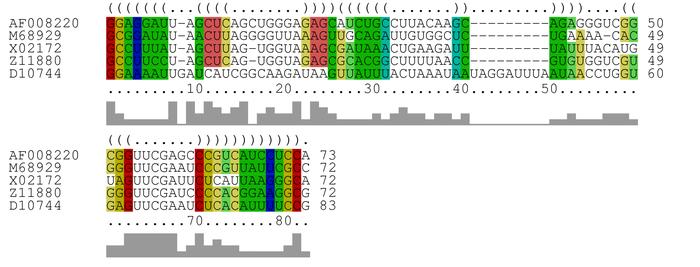
CARNA
CARNA
is a tool for multiple alignment of RNA molecules. CARNA requires only the
RNA sequences as input and will compute base pair probability matrices and
align the sequences based on their full ensembles of structures.
Alternatively, you can also provide base pair probability matrices (dot
plots in .ps format) or fixed structures (as annotation in the FASTA
alignment) for your sequences. If you provide fixed structures, only those
structures and not the entire ensemble of possible structures is aligned.
In contrast to LocARNA, CARNA does not pick the most likely consensus
structure, but computes the alignment that fits best to all likely
structures simultaneously. Hence, CARNA is particularly useful when
aligning RNAs like riboswitches, which have more than one stable
structure.
(Reference: A. Dragos et al. 2012. Nucleic Acids
Reseach 40: W49-W53).
Foldalign
Foldalign
- folds and aligns RNA structures (make a foldalignment) based on a
lightweight energy model and sequence similarity. The current version
makes pairwise fold alignments.
(Reference: J. H. Havgaard et al. (2007) J. PLOS
computational biology. 3: e193).
MFold
For RNA folding use
MFold
- N.B. The data can be presented in a number of graphic formats. This is
my "go to" site if I'm interested in a secondardy structure for a fragment
of RNA or DNA
(Reference: M. Zuker. 2003. Nucleic Acids Res. 31:
3406-3415).
Vienna RNA secondary structure prediction
Vienna RNA secondary structure prediction (University of Vienna, Austria). I have found this site useful for drawing tRNAs in cloverleaf format.
CONTRAfold
CONTRAfold
is a novel secondary structure prediction method based on conditional
log-linear models, a flexible class of probabilistic models which
generalize upon stochastic context-free grammars by using discriminative
training and feature-rich scoring. By incorporating most of the features
found in typical thermodynamic models, CONTRAfold achieves the highest
single sequence prediction accuracies to date, outperforming currently
available probabilistic and physics-based techniques. It provides
MARNA-like output couples with hairpin structures
(Reference: Do, C.B. et al. 2006. Bioinformatics 22:
e90-e98).
Web-Beagle
Web-Beagle -
a web server for the pairwise global or local alignment of RNA secondary
structures.
(Reference: E. Mattei et al. 2015. Nucl. Acids Res.
43 (W1): W493-W497).
Rclick
Rclick
this web server that is capable of superimposing RNA 3D structures by
using clique matching and 3D least-squares fitting. Rclick has been
benchmarked and compared with other popular servers and methods for RNA
structural alignments. In most cases, Rclick alignments were better in
terms of structure overlap. It also recognizes conformational changes
between structures.
(Reference: Nguyen MN, & Verma C. 2015.
Bioinformatics 31:966-968).
RNAComposer
RNAComposer -
is a fully automated RNA structure modeling server
(Reference: Biesiada M et al. 2016. Methods 103: 120 - 127).
RNAtive
RNAtive
- is a consensus-based RNA structure analysis system designed to process multiple structural models sharing the same
sequence and to identify reliable base pairs and stacking interactions. It supports model validation, improves structural
predictions, and facilitates studies of RNA structure evolution. The tool accepts a minimum of two RNA 3D structure models
in PDB or mmCIF format (with a total file size limit of 100 MB), and analyzes them using state-of-the-art base-pair annotation
tools.
(Reference: Pielesiak J et al. 2025. Bioinformatics 41(11): btaf601).
RNAstructure
RNAstructure
(webservers for RNA secondary structure prediction) is a software package
that includes structure prediction by free energy minimization, prediction
of base pairing probabilities, prediction of structures composed of highly
probably base pairs, and prediction of structures with pseudoknots.
(Reference: Xu ZZ & Mathews DH (2016) Methods Mol
Biol; 1490: 15-34).
RNAfold
RNAfold - this web server will predict secondary structures of single stranded RNA or DNA sequences. Current limits are 7,500 nt for partition function calculations and 10,000 nt for minimum free energy only predictions.
Vfold Pipeline
Vfold Pipeline
- offers a new user-friendly approach to the fully automated prediction of
RNA 3D structures with given sequences. It first predicts 2D structures
using the Vfold2D model and then predicts 3D structures based on the
predicted 2D structures using the Vfold3D and VfoldLA models.
(Reference: Xu, XJ., Chen, S.-J. (2016) Methods Mol
Biol. 1490: 63-72).
LocARNA
LocARNA
- Multiple Alignment of RNAs - is a tool for multiple alignment of RNA
molecules. LocARNA requires only RNA sequences as input and will
simultaneously fold and align the input sequences. LocARNA outputs a
multiple alignment together with a consensus structure. For the folding it
makes use of a very realistic energy model for RNAs as it is by RNAfold
of the Vienna RNA package (or Zuker's mfold). For the alignment it
features RIBOSUM-like similarity scoring and realistic gap cost.
(Reference: C. Smith et al. 2010. Nucl. Acids Res.
38: W373-377).
CARNA
CARNA
is a tool for multiple alignment of RNA molecules. CARNA requires only the
RNA sequences as input and will compute base pair probability matrices and
align the sequences based on their full ensembles of structures.
Alternatively, you can also provide base pair probability matrices (dot
plots in .ps format) or fixed structures (as annotation in the FASTA
alignment) for your sequences. If you provide fixed structures, only those
structures and not the entire ensemble of possible structures is aligned.
In contrast to LocARNA, CARNA does not pick the most likely consensus
structure, but computes the alignment that fits best to all likely
structures simultaneously. Hence, CARNA is particularly useful when
aligning RNAs like riboswitches, which have more than one stable
structure.
(Reference: A. Dragos et al. 2012. Nucleic Acids
Reseach 40: W49-W53).
Foldalign
Foldalign
- folds and aligns RNA structures (make a foldalignment) based on a
lightweight energy model and sequence similarity. The current version
makes pairwise fold alignments.
(Reference: J. H. Havgaard et al. (2007) J. PLOS
computational biology. 3: e193).
MFold
For RNA folding use
MFold
- N.B. The data can be presented in a number of graphic formats. This is
my "go to" site if I'm interested in a secondardy structure for a fragment
of RNA or DNA
(Reference: M. Zuker. 2003. Nucleic Acids Res. 31:
3406-3415).
Vienna RNA secondary structure prediction
Vienna RNA secondary structure prediction (University of Vienna, Austria). I have found this site useful for drawing tRNAs in cloverleaf format.
CONTRAfold
CONTRAfold
is a novel secondary structure prediction method based on conditional
log-linear models, a flexible class of probabilistic models which
generalize upon stochastic context-free grammars by using discriminative
training and feature-rich scoring. By incorporating most of the features
found in typical thermodynamic models, CONTRAfold achieves the highest
single sequence prediction accuracies to date, outperforming currently
available probabilistic and physics-based techniques. It provides
MARNA-like output couples with hairpin structures
(Reference: Do, C.B. et al. 2006. Bioinformatics 22:
e90-e98).
Web-Beagle
Web-Beagle -
a web server for the pairwise global or local alignment of RNA secondary
structures.
(Reference: E. Mattei et al. 2015. Nucl. Acids Res.
43 (W1): W493-W497).
Rclick
Rclick
this web server that is capable of superimposing RNA 3D structures by
using clique matching and 3D least-squares fitting. Rclick has been
benchmarked and compared with other popular servers and methods for RNA
structural alignments. In most cases, Rclick alignments were better in
terms of structure overlap. It also recognizes conformational changes
between structures.
(Reference: Nguyen MN, & Verma C. 2015.
Bioinformatics 31:966-968).
RNAComposer
RNAComposer -
is a fully automated RNA structure modeling server
(Reference: Biesiada M et al. 2016. Methods 103: 120 - 127).
RNAtive
RNAtive
- is a consensus-based RNA structure analysis system designed to process multiple structural models sharing the same
sequence and to identify reliable base pairs and stacking interactions. It supports model validation, improves structural
predictions, and facilitates studies of RNA structure evolution. The tool accepts a minimum of two RNA 3D structure models
in PDB or mmCIF format (with a total file size limit of 100 MB), and analyzes them using state-of-the-art base-pair annotation
tools.
(Reference: Pielesiak J et al. 2025. Bioinformatics 41(11): btaf601).
Pseudoknots:
pKiss
pKiss
- is the successor of pknotsRG, the first pseudoknot class is the
canonical simple recursive pseudoknot from pknotsRG. The new class are
canonical simple recursive kissing hairpins.
(Reference: Janssen, S. & Giegerich, R.
Bioinformatics, 2015; 31(3):423-5).
ProbKnot
ProbKnot
server takes a sequence file of nucleic acids, either DNA or RNA, and
predicts the presence of pseudoknots in its folded configuration. Note
that increasing the number of calculation iterations may be helpful in
increasing accuracy. Note also that if a pseudoknot-containing structure
is predicted, it will be displayed as a circular structure. If the
predicted structure does not contain pseudoknots, it will be displayed as
a radial structure
(Reference: Bellaousov S, Mathews DH (2010) RNA.
16(10): 1870-1880).
CyloFold
CyloFold
(part of RNA Structure and Design Tools) - is a program for predicting the
secondary structure of an RNA sequence including pseudoknots. For
sequences greater 100 nucleotides, it is recommended to save the provided
job ID, in order to be able to access the status of the job submission.
Once the computation is finished, the predicted secondary structure is
displayed in two different formats ("bracket-notation" as well as
"CT-format") or
here.
(Reference: Bindewald E, Kluth T, Shapiro BA (2010)
Nucleic Acids Res. 38(Web Server issue): W368-372)
vsfold5
vsfold5 - RNA Pseudoknot Prediction Server (requires
registration)
GCGGCCAGCUCCAGGCCGCCAAACAAUAUGGAGCAC
((((((..[[[[[)))))).........]]]]]...
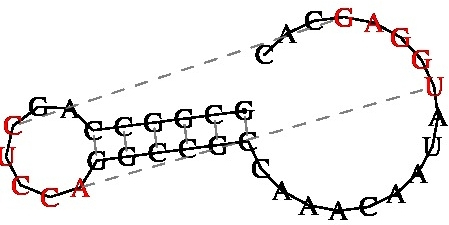
KineFold Web Server
KineFold Web Server
- RNA/DNA folding predictions including pseudoknots and entangled helices
(Reference: A. Xayaphoummine et al. (2005) Nucleic
Acid Res. 33: 605-610).
IPknot
IPknot:
IP-based prediction of RNA pseudoKNOTs - rovides services for predicting
RNA secondary structures including a wide class of pseudoknots. IPknot can
also predict the consensus secondary structure when a multiple alignment
of RNA sequences is given.
(Reference: K. Sato et al. (2011) Bioinformatics, 27:
i85-i93).
ProbKnot
ProbKnot
- this server takes a sequence file of nucleic acids, either DNA or RNA,
and predicts the presence of pseudoknots in its folded configuration. Note
that increasing the number of calculation iterations may be helpful in
increasing accuracy. Note also that if a pseudoknot-containing structure
is predicted, it will be displayed as a circular structure. If the
predicted structure does not contain pseudoknots, it will be displayed as
a radial structure.
(Reference: Bellaousov S, Mathews DH (2010) RNA.
16(10): 1870-1880).
RNAstructure - Predict a Secondary Structure Web Server
RNAstructure - Predict a Secondary Structure Web Server - combines many separate prediction and analysis algorithms: calculating a partition function, predicting a maximum free energy (MFE)structure, finding structures with maximum expected accuracy, and pseudoknot prediction. This servertakes a sequence, either RNA or DNA, and creates ahighly probable, probability annotated group of secondary structures, starting with the lowest free energy structure and including others with varied probabilities of correctness.
K2N
K2N:
a service to get from knotted to nested RNA structures. This site provides
access to a variety of methods for pseudoknot removal.
(Reference: S. Smit et al. (2008) RNA 14(3):410-416).
Other regulatory elements:
Riboswitch Finder
Riboswitches are structured noncoding RNA domains used by many bacteria to
monitor the concentrations of target ligands and regulate gene expression
accordingly (See:
Discovering riboswitches: the past and the future
and
Parallel Discovery Strategies Provide a Basis for Riboswitch Ligand Design).
They can be discovered using
Riboswitch Finder
(Reference: Bengert P, & Dandekar T. (2004) Nucleic
Acids Res. 32(Web Server issue): W154-W159)
RNAProbe
RNAProbe - facilitates normalization, analysis, and visualization
of the low-pass SHAPE, DMS and CMCT probing results with the modification sites detected by capillary electrophoresis. It
performs normalization based on a well-established protocol, utilizes recognized secondary structure prediction methods,
and generates high-quality images with structure representations and reactivity heatmaps.
(Reference: Wirecki TK et al. (2020) Nucleic Acids
Res. 48(Issue W1): W292-W299).
SiRNA:
ARTS
ARTS
(Alignment of RNA Tertiary Structures) - aligns two nucleic acid
structures (RNAs or DNAs) in pdb format and detecting apriori unknown
common substructures. The identified common substructures can be either
large global folds or small local tertiary motifs with at least two
successive base pairs.
(Reference: Dror O et al. 2005. Bioinformatics 21
(Suppl 2):ii47-ii53)
CopraRNA
CopraRNA
is a tool for sRNA target prediction. It computes whole genome predictions
by combination of distinct whole genome IntaRNA predictions
(Reference: Wright PR et al. 2014. Nucl. Acids Res.
42 (W1), W119-W123).
OligoWalk
OligoWalk
- calculates thermodynamic features of sense-antisense hybidization. It
predicts the free energy changes of oligonucleotides binding to a target
RNA. It can be used to design efficient siRNA targeting a given mRNA
sequence.
(Reference: Lu ZJ & Mathews DH. 2008. Nucleic
Acids Res.36: 640-647).
CRISPR:
CRISPR sgRNA Design Tool
CRISPR sgRNA Design Tool - GenScript is proud to offer free online access to our gRNA sequence design tool, developed by the Broad Institute of Harvard and MIT. Our gRNA design tool will identify single guide RNAs for use with wild-type S. pyogenes Cas9 for any DNA sequence you input. Start your gRNA design project by entering a sequence up to 250bp in length below.
CRISPRdirect
CRISPRdirect - is a
simple and functional web server for selecting rational CRISPR/Cas targets
from an input sequence.
(Reference: Naito Y et al. (2015)Bioinformatics 31:
1120-1123).
Center for Non-coding RNA in Technology and Health (RTH, Denmark)
The Center for Non-coding RNA in Technology and Health (RTH, Denmark) has developed CRISPRon - a CRISPR-Cas9 guide efficiency prediction server; WebCircRNA - assesses the circular RNA potential of coding and noncoding RNA; CRISPRoff - off-targeting assessment of Cas9 gRNAs plus a number of other sites for ncRNA detection and analysis.
META SITE:
Rtools
Rtools (Computational Biology Research Consortium , Japan) - is a bioinformatics web Server for RNA (single FASTA format, <= 400nt) - In order to visualize the whole picture of the distribution of the secondary structure, this web-server provides users with rich information of single RNA sequences using 7 tools: (a) CentroidFold based on a generalized centroid estimator is one of the most accurate tools for predicting RNA secondary structures. (b) CentroidHomfold predicts RNA secondary structures by employing automatically collected homologous sequences of the target. (c) IPknot predicts RNA secondary structures including a wide class of pseudoknots. (d) Rchange computes entropy and internal energy changes of secondary structures for single-point mutated sequences. (e) CapR calculates probabilities that each RNA base position is located within each secondary structural context for long RNA sequences. (f) Raccess computes the accessibility of segment [a, b] = [x, x+l-1] in the transcript for all the positions x with fixed length l (Acc.len) = 5, 10, 20. (g) RintD validates RNA secondary structures. Target secondary structures are predicted by CentroidFold (inference engine: McCaskill) and RNAfold (Minimum free energy structure).
Updated: February, 2026